
Drug Discovery using AI Full Syllabus

Module 1: Introduction to AI in Drug Discovery

Traditional vs AI-Based Drug Discovery
Drug discovery has historically been a long, expensive, and uncertain process. Traditional approaches rely heavily on trial-and-error methods, high-throughput screening, and years of laboratory research. It often takes more than a decade and billions of dollars to bring a single drug to market, with a high risk of failure at different stages.
AI-based drug discovery changes this approach by using advanced algorithms, machine learning, and data-driven insights to accelerate the entire process. Instead of screening millions of compounds manually, AI can analyze huge datasets, predict drug-target interactions, and shortlist the most promising candidates in a fraction of the time. This not only speeds up research but also improves decision-making for scientists.
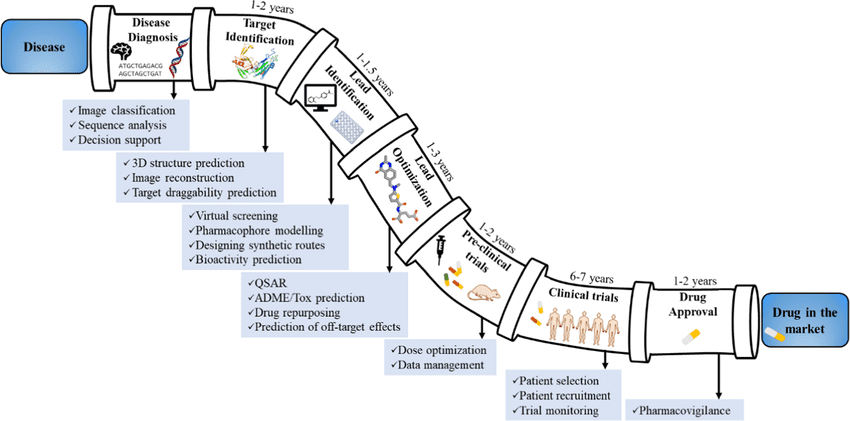
Overview of the Drug Development Lifecycle
The drug development lifecycle typically involves several stages:
- Target Identification – Finding a biological target such as a protein or gene associated with a disease.
- Hit Discovery – Screening chemical compounds that might interact with the target.
- Lead Optimization – Modifying promising compounds to improve effectiveness and reduce side effects.
- Preclinical Testing – Evaluating safety and efficacy in laboratory and animal studies.
- Clinical Trials – Testing in human participants across multiple phases to confirm safety, dosage, and effectiveness.
- Regulatory Approval and Launch – Gaining approval from authorities before making the drug available to patients.
Each of these stages is critical, and delays or failures at any step can lead to huge losses in time and money.
Where AI Fits In
AI is integrated across the drug discovery pipeline to make the process more efficient and accurate.
- Target Identification: AI helps analyze genetic, proteomic, and biomedical data to identify potential disease-causing targets.
- Hit and Lead Discovery: Machine learning models predict which compounds are likely to bind effectively with the target.
- Optimization: AI tools can simulate molecular interactions and suggest chemical modifications to improve drug performance.
- Clinical Trials: AI assists in patient selection, predicting trial outcomes, and reducing dropout rates by identifying ideal trial candidates.
Key Benefits of AI in Drug Discovery
- Time-Saving: Reduces years of research to months by quickly analyzing vast data.
- Cost Reduction: Minimizes the need for repetitive lab experiments, lowering overall expenses.
- Higher Accuracy: Improves the prediction of drug-target interactions, reducing trial failures.
- Scalability: Allows researchers to explore millions of compounds efficiently.
- Personalization: Supports the development of precision medicines tailored to individual patients.
Module 2: Biology & Chemistry Basics (For AI Learners)
DNA, RNA, and Proteins: The Foundation of Life
Every living organism is built on the blueprint of DNA, which stores genetic information. DNA is transcribed into RNA, and RNA is translated into proteins. Proteins are the workhorses of the cell, responsible for carrying out vital functions such as signaling, defense, and metabolism.
For AI learners, it is important to understand that diseases often occur when something goes wrong in this flow of information — for example, a mutation in DNA or a misfolded protein. Drug discovery often aims to correct or influence these processes.
Receptors and Enzymes in Drug Targeting
Proteins can act as receptors or enzymes, and both are critical in drug discovery.
- Receptors are protein molecules on the surface of cells that receive signals, like a lock that only fits a specific key. Drugs can act as that key to either activate (agonists) or block (antagonists) the receptor.
- Enzymes are proteins that catalyze biochemical reactions in the body. Many diseases are caused by overactive or malfunctioning enzymes. Drugs designed to inhibit or enhance enzyme activity can help restore balance.
AI models use structural data of receptors and enzymes to predict how different molecules will interact with them, which is a cornerstone of modern drug discovery.
Small Molecules vs Biologics
Drugs generally fall into two broad categories: small molecules and biologics.
- Small molecules are low molecular weight compounds, often taken as pills. They can easily enter cells and interact with intracellular targets. Classic examples include aspirin and antibiotics.
- Biologics are larger, complex molecules such as antibodies, peptides, or gene therapies. They are usually administered by injection and are designed to target very specific biological processes. For example, monoclonal antibodies are widely used in cancer and autoimmune disease treatment.
For AI in drug discovery, the distinction matters because predicting interactions of small molecules is very different from modeling the behavior of large biologics.
Chemical Properties and Molecular Structures
At the heart of chemistry in drug discovery is understanding how the structure of a molecule affects its function. Key chemical properties include:
- Solubility – how well a drug dissolves in the body.
- Stability – how long a compound remains active without breaking down.
- Binding Affinity – how strongly a molecule binds to its target.
- Selectivity – whether a drug affects only the intended target or also interacts with other molecules (leading to side effects).
Molecular structures can be represented in 2D or 3D. AI uses these structural representations to predict drug-likeness, toxicity, and interactions. Machine learning algorithms, for example, can analyze chemical fingerprints or molecular graphs to quickly assess whether a compound is worth pursuing.
Module 3: Types of Data in Drug Discovery
Genomic and Proteomic Data
Drug discovery today relies heavily on understanding the human genome and proteome. Genomic data reveals variations in DNA that may be linked to diseases, while proteomic data shows how proteins are expressed, folded, and modified. Since most drugs act on proteins, this data is critical for identifying disease targets and predicting how a treatment might work.
Molecular Compound Libraries (SMILES, InChI)
Researchers use large digital libraries of molecules to explore potential drug candidates. Two common ways of representing these compounds are:
- SMILES (Simplified Molecular Input Line Entry System): a text string that encodes a chemical structure in a simple, linear form.
- InChI (International Chemical Identifier): a more detailed and standardized way of representing molecules for database searches and sharing.
These libraries allow AI algorithms to quickly process millions of compounds and identify the most promising ones for further testing.
Bioactivity Data (IC50, EC50, etc.)
Not all compounds behave the same way. Bioactivity data measures how strongly or weakly a molecule interacts with a target. Common measures include:
- IC50: the concentration of a compound needed to inhibit a biological process by 50%.
- EC50: the concentration needed to produce 50% of the maximum effect.
This data helps distinguish between strong drug candidates and weak or inactive compounds.
Key Data Sources: PubChem, ChEMBL, DrugBank, BindingDB
Public databases play a huge role in AI-driven drug discovery:
- PubChem: a vast resource of chemical structures and their properties.
- ChEMBL: focused on bioactivity data, connecting compounds with their biological effects.
- DrugBank: integrates detailed drug information, including mechanisms and targets.
- BindingDB: specializes in protein-ligand binding data.
AI systems tap into these sources to train predictive models that can forecast drug behavior more accurately.
Module 4: Data Preparation & Feature Engineering
Molecular Fingerprinting (MACCS, ECFP)
Just like humans use fingerprints for identification, molecules also have “fingerprints.” These are unique digital codes representing molecular features.
- MACCS Keys: a set of predefined chemical substructures used as binary fingerprints.
- ECFP (Extended Connectivity Fingerprints): circular fingerprints that capture detailed information about a molecule’s structure.
Fingerprints make it easier for AI models to compare compounds and predict similarities.
One-Hot Encoding for Sequences
When dealing with sequences like DNA, RNA, or protein chains, AI models need a numerical representation. One-hot encoding turns each base (A, T, G, C) or amino acid into a binary vector. For example, “A” becomes [1,0,0,0]. This helps algorithms process sequence data without losing biological meaning.
Descriptor Calculation (Molecular Weight, logP)
Descriptors are numerical values that summarize key properties of a molecule:
- Molecular Weight: helps predict how the drug will be absorbed and distributed.
- logP (Partition Coefficient): measures how soluble a molecule is in fat vs water, which affects absorption and bioavailability.
Descriptors act as input features for AI models, enabling them to predict drug-likeness and behavior.
Handling Imbalanced Datasets (Active vs Inactive Compounds)
In drug discovery, most compounds in a dataset are inactive, with only a small fraction being active. This imbalance can mislead AI models into always predicting “inactive.” To fix this, techniques such as oversampling actives, undersampling inactives, or using cost-sensitive learning are applied. Balancing the dataset ensures that the model does not overlook potential drug candidates.
Module 5: Machine Learning Models for Drug Discovery

Classification Models for Compound Activity Prediction
In drug discovery, one of the most important questions is: “Will this compound be active or inactive?” Classification models help answer this by labeling compounds based on their biological activity. For example, a model might predict whether a molecule inhibits a cancer-related enzyme or not. This helps researchers filter through thousands of possibilities and focus only on promising candidates.
Regression Models for Drug–Target Binding Affinity
Sometimes, it’s not enough to know whether a compound is active — researchers also need to measure how strong the interaction is between a drug and its target protein. This is where regression models come in. Instead of predicting categories, they predict numerical values such as binding affinity (how tightly a drug binds) or IC50/EC50 values. Strong binding usually means a more effective drug, so regression helps in fine-tuning potential candidates.
Popular Algorithms: Random Forest, SVM, and XGBoost
Several machine learning algorithms are widely used in analyzing drug datasets:
- Random Forest (RF): A collection of decision trees that can classify compounds and handle noisy data well. It’s known for stability and interpretability.
- Support Vector Machines (SVMs): Effective for high-dimensional data, SVMs are commonly used when datasets include thousands of molecular descriptors. They excel at separating active vs inactive compounds.
- XGBoost: A powerful boosting algorithm that often outperforms others in predictive accuracy. It is especially good for large, imbalanced datasets in drug discovery.
Each of these algorithms has strengths, and researchers often experiment with multiple models to see which performs best for a given dataset.
Model Interpretability in Biomedical Research
In drug discovery, it’s not enough for a model to make accurate predictions — scientists also need to understand why the model made those predictions. Interpretability ensures that results can be trusted and validated in real-world experiments.
Techniques such as feature importance analysis (e.g., which molecular property influenced the prediction most) or SHAP values (explaining individual predictions) help researchers link machine learning outputs to biological reasoning. This builds confidence in using AI as a decision-support tool in biomedical research.
Module 6: Deep Learning for Molecule Generation & Prediction
Convolutional Neural Networks (CNNs) for 2D/3D Molecular Image Analysis
CNNs, originally developed for image recognition, are powerful tools for drug discovery. Molecules can be represented as 2D chemical diagrams or 3D structural models, much like images. CNNs can “see” patterns such as functional groups, bonds, or spatial arrangements and use these features to predict biological activity.
For example, a CNN might learn to identify structural motifs that are often linked with cancer-inhibiting properties. This makes them useful in classifying molecules or screening large compound libraries quickly.
Recurrent Neural Networks (RNNs) for SMILES Sequence Generation
Molecules can also be described as text strings using SMILES notation. RNNs, which are designed to process sequential data, can learn the “grammar” of chemistry. By training on millions of SMILES strings, an RNN can generate entirely new molecules that follow chemical rules but may never have been synthesized before.
This generative approach allows AI to suggest novel compounds that could become future drugs, cutting down on blind trial-and-error.
Autoencoders for Molecule Optimization
Autoencoders are neural networks that compress data into a smaller representation and then reconstruct it. In drug discovery, autoencoders can learn the latent space of molecules—essentially the hidden patterns that define chemical behavior.
By manipulating this latent space, researchers can optimize molecules for properties such as solubility, potency, or reduced toxicity. For instance, if a molecule is effective but poorly absorbed, autoencoders can suggest modifications that improve absorption without losing effectiveness.
Graph Neural Networks (GNNs) for Molecular Graphs
Molecules are naturally structured as graphs: atoms are nodes, and bonds are edges. GNNs are specifically designed to work with such graph data. Unlike CNNs or RNNs, which need molecules to be converted into images or strings, GNNs work directly on molecular graphs.
This allows them to capture the true connectivity and relationships between atoms. GNNs can predict drug-target interactions, estimate toxicity, and even design new molecules by “reasoning” over the graph structure.
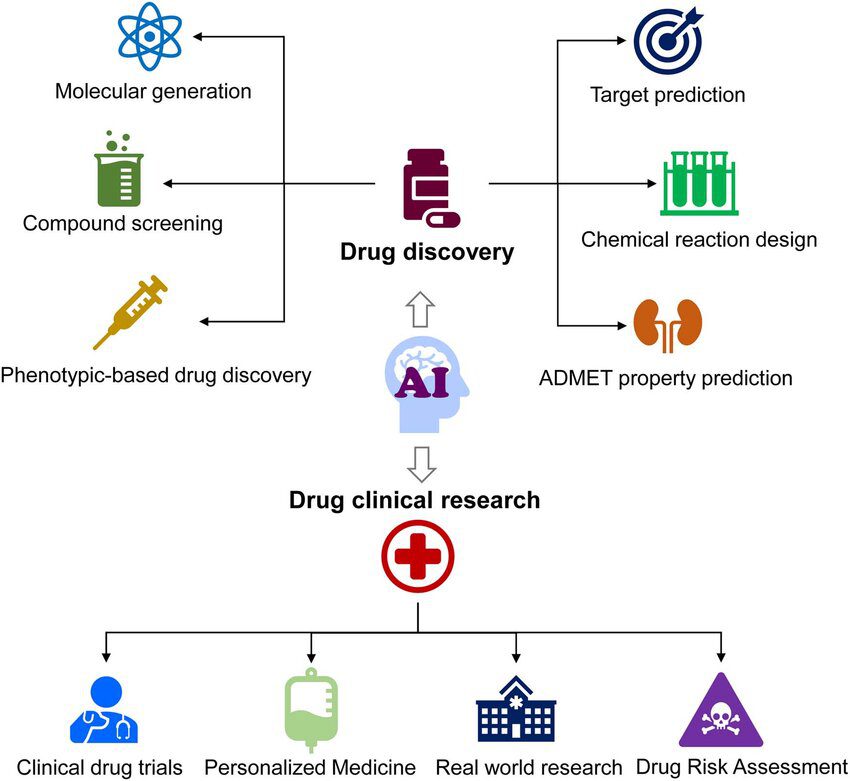
Module 7: AI in Target Identification & Validation& De Novo Drug Design
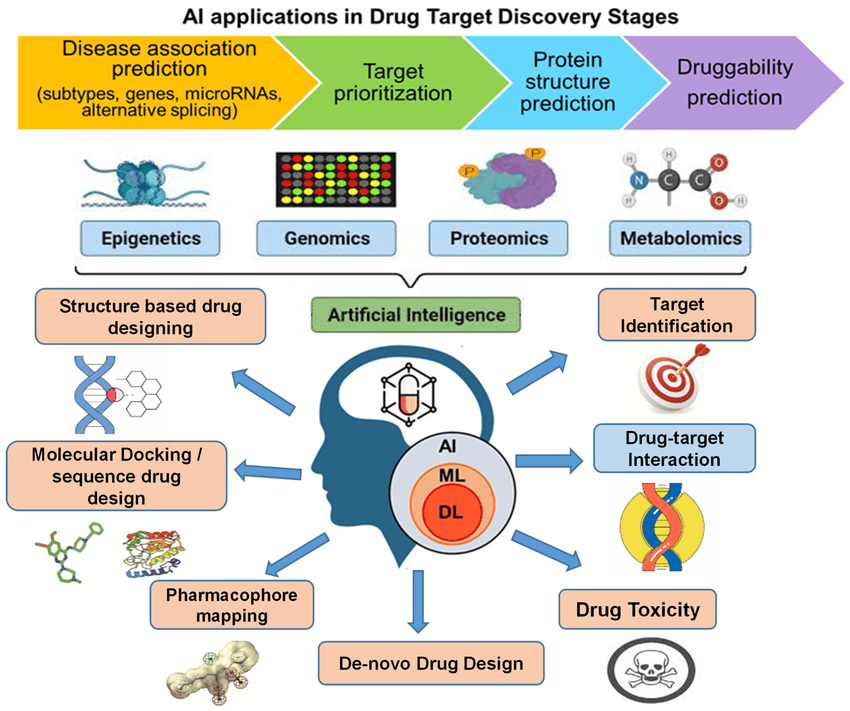
AI in Target Identification & Validation
Before a drug can be created, scientists must understand where and how the drug will act in the human body. This is where AI plays a powerful role:
- Disease Gene Mapping with Machine Learning
AI analyzes large genomic and proteomic datasets to identify genes linked to diseases. By spotting hidden patterns, it helps researchers pinpoint which genes or proteins may be driving an illness. - Predicting Protein–Drug Interactions
Traditionally, testing protein–drug binding required expensive lab experiments. AI models can now predict how a potential drug molecule interacts with a target protein, saving both time and resources. - Docking Score Prediction with AI
Molecular docking estimates how strongly a drug binds to its target. AI-driven models can predict docking scores with higher speed and accuracy compared to classical simulations. - Popular Tools
Platforms like DeepChem, AlphaFold, and BindingDB allow researchers to explore protein structures, test drug interactions, and validate targets with AI-powered algorithms.
AI in De Novo Drug Design
Once a target is identified and validated, the next step is designing completely new drug molecules. AI brings creativity and intelligence into this process:
- Generative Models (GANs, VAEs)
Just like AI can generate art or music, Generative Adversarial Networks (GANs) and Variational Autoencoders (VAEs) can create novel chemical structures. These molecules are designed to potentially bind well with the target protein. - Reinforcement Learning for Compound Optimization
AI agents are trained to explore the chemical space, learning from feedback signals. For example, if a generated molecule binds strongly or shows better drug-like features, the model “rewards” itself and keeps improving. - Synthetic Accessibility & Drug-Likeness Prediction
A molecule may look promising on paper but must also be easy to synthesize in the lab. AI evaluates whether a molecule is practical to create and predicts its drug-like properties, such as solubility and stability. - Toxicity Filtering with AI
Safety is a top priority in drug development. AI screens candidate molecules early on, filtering out those with toxic side effects, which reduces the risk of costly failures in later clinical trials.
Why These Modules Matter
Together, Target Identification (Module 7) and De Novo Drug Design (Module 8) represent the core of AI-driven drug discovery. By combining data-driven insights with generative creativity, AI accelerates the journey from understanding disease mechanisms to producing safe and effective drug candidates.
This integration not only saves years of research and millions of dollars, but also increases the chances of success in creating breakthrough medicines.
Module 8: Advanced Applications of AI in Drug Discovery: From Virtual Screening to Future Trends
Artificial Intelligence (AI) has already transformed the earliest stages of drug discovery, such as target identification and molecule design. However, the true power of AI is revealed when it is applied across the entire drug development pipeline—from virtual screening of compounds to clinical trial predictions, and even shaping future policies and ethics. This module combines the advanced concepts from virtual screening, predictive modeling, clinical applications, ethical issues, and hands-on projects into one integrated chapter.
1. Virtual Screening Using AI
Traditionally, drug candidates were tested one by one in laboratories, a process that consumed both time and billions of dollars. Virtual screening (VS) revolutionizes this by allowing researchers to computationally test millions of molecules before stepping into a lab. AI takes this process a step further by making it faster, more accurate, and scalable.
- Ligand-Based vs. Structure-Based Screening
- Ligand-based screening compares new molecules with known active compounds, predicting activity based on similarity.
- Structure-based screening relies on the 3D structure of proteins and predicts how candidate molecules can bind to them. AI enhances both methods by spotting hidden chemical features that traditional methods often miss.
- Machine Learning-Assisted Scoring & Ranking
After generating possible interactions, AI models can quickly score and rank thousands of molecules to highlight the ones most likely to succeed in wet-lab experiments. Models such as XGBoost and deep neural networks outperform conventional scoring functions by learning from massive historical datasets. - ADMET Property Prediction
A molecule’s success doesn’t only depend on binding to a protein; it also depends on how the body processes it. AI predicts Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties early in the pipeline, reducing late-stage failures. - Popular Tools
Platforms like PyRx, Deep Docking, and Mol2Vec are widely used for AI-based screening. These tools integrate machine learning with molecular docking simulations, making it possible to virtually evaluate millions of compounds in days rather than years.
2. AI in Clinical Trials & Predictive Modeling
Even if a molecule passes screening and pre-clinical testing, the real test lies in clinical trials—a stage notorious for high failure rates. AI can transform this stage by making trials smarter, safer, and more efficient.
- Patient Selection Models Using Genomics
One of the leading reasons for trial failure is enrolling the wrong patients. AI models analyze genomic and clinical data to select patients who are most likely to respond positively to the treatment. - Predicting Trial Success or Failure
By studying patterns from past clinical trials, machine learning models can predict the probability of a trial succeeding or failing before it begins. This saves billions by avoiding high-risk investments. - Integration with Electronic Health Records (EHRs)
AI can mine EHRs to find real-world patient data that supports trial design. This creates more personalized trials that reflect actual clinical environments rather than narrow, controlled studies. - Case Study: AI in COVID-19 Drug Repurposing
During the pandemic, AI played a major role in identifying existing drugs that could be repurposed to treat COVID-19. By screening massive drug libraries against viral proteins, AI tools proposed candidates like Remdesivir and Baricitinib, dramatically speeding up research.
3. AI Tools & Platforms in Drug Discovery
The AI revolution in pharma is supported by a wide ecosystem of libraries, platforms, and datasets. Mastering these tools is critical for researchers entering this space.
- Cheminformatics Libraries
- RDKit: Widely used for molecule representation, fingerprinting, and descriptor calculations.
- Open Babel: Helps convert molecular file formats and analyze chemical structures.
- AI Platforms
- DeepChem: Open-source platform offering pre-built AI models for chemistry and biology.
- Chemprop: A graph neural network tool designed for molecular property prediction.
- IBM RXN: Cloud-based platform for predicting chemical reactions and synthesis pathways.
- Insilico Medicine: A commercial AI drug discovery platform known for pioneering de novo design.
- Datasets
Training AI models requires high-quality datasets. Popular sources include:- ZINC: A database of purchasable compounds for screening.
- Tox21: A dataset for predicting chemical toxicity.
- DrugBank: Combines detailed drug information with molecular data.
4. Regulatory, Ethical & Future Trends in AI Drug Discovery
While AI offers immense promise, the field must also confront challenges around regulation, ethics, and fairness.

- FDA Guidance for AI-Based Drug Models
Regulatory agencies like the FDA and EMA are working on frameworks to evaluate AI models. Transparency, reproducibility, and interpretability are essential for approval. - Transparency & Explainability
A major challenge in deep learning is the “black box” nature of models. In drug discovery, explainability is not optional—researchers and regulators need to understand why an AI recommends a certain molecule. - Bias in AI Models
If datasets are biased (e.g., under-representing certain populations), then the resulting drugs may not work equally well for everyone. Ensuring fairness in drug discovery is an ethical necessity. - Future Trends: Quantum Machine Learning (QML)
The future may combine AI with quantum computing to simulate molecular interactions at an atomic level with unprecedented precision. This could unlock completely new classes of medicines.
5. Capstone Project (Hands-On Learning)
To consolidate learning, a hands-on project is highly recommended.
Project Idea: Predict Binding Affinity of Small Molecules
- Step 1: Select a protein target (e.g., a cancer-related enzyme).
- Step 2: Collect a dataset from DrugBank or BindingDB.
- Step 3: Preprocess molecules using RDKit (generate fingerprints, calculate descriptors).
- Step 4: Train ML/DL models using Scikit-learn or TensorFlow to predict binding affinity.
- Step 5: Evaluate results and visualize using Matplotlib or Plotly.
- Step 6: Propose the top candidate molecules for further testing.
This project mirrors real-world drug discovery workflows and prepares learners for industry-level tasks.
6. Programming Languages & Tools
AI in drug discovery is heavily Python-driven, and learners should be comfortable with the following ecosystem:
- Python Libraries: RDKit, DeepChem, Scikit-learn, TensorFlow, PyTorch
- Environments: Jupyter Notebook or Google Colab for interactive coding
- Visualization: Seaborn, Matplotlib, Plotly for clear scientific graphics

AI is no longer a supporting tool—it is becoming the backbone of drug discovery. From virtual screening and predictive modeling to clinical trials and ethical frameworks, AI accelerates the journey of a molecule from computer to clinic. The integration of cutting-edge tools, ethical practices, and future innovations like quantum machine learning will define the next era of personalized and efficient medicine.
This combined module equips learners with both theoretical foundations and practical skills to contribute meaningfully to the AI-driven revolution in healthcare.

